
Associate members: Andrea Cavalli
Status: In progress
Alzheimer’s disease (AD) is recognized as the most spread neurodegenerative disease affecting over 30 million people worldwide. The development of the disorder has been linked to the presence of extracellular beta-amyloid (Ab) peptide aggregates of different sizes. Ab oligomeric species formed at early stages of the aggregation process are leading candicates for causing AD. Thus, targeting oligomers can be a very valuable strategy to combat Alzheimer’s disease. However, the molecular mechanism underlying the self-assembly of the different Ab species is not fully understood and it is not clear how the early soluble oligomeric species associate to form protofibrils and, subsequently, mature fibrils.
The main objective of this project is to apply computational techniques to elucidate small angle X-ray scattering (SAXS) data collected by our collaborators at University of Cambridge (Prof. M. Vendruscolo group) and resolve major coexisting components in Ab fibrillation process.
Beltrandi, M., et al., Insights into the coiled-coil organization of the Hendra virus phosphoprotein from combined biochemical and SAXS studies. Virology, 2015. 477C: p. 42-55.

Nipah and Hendra viruses are recently emerged paramyxoviruses belonging to the Henipavirus genus. The Henipavirus phosphoprotein (P) consists of a large intrinsically disordered domain and a C-terminal domain (PCT) containing alternating disordered and ordered regions. Among these latter is the P multimerization domain (PMD). Using biochemical, analytical ultracentrifugation and small-angle X-ray scattering (SAXS) studies, we show that Hendra virus (HeV) PMD forms an elongated coiled-coil homotrimer in solution, in agreement with our previous findings on Nipah virus (NiV) PMD. However, the orientation of the N-terminal region differs from that observed in solution for NiV PMD, consistent with the ability of this region to adopt different conformations. SAXS studies provided evidence for a trimeric organization also in the case of PCT, thus extending and strengthening our findings on PMD. The present results are discussed in light of conflicting reports in the literature pointing to a tetrameric organization of paramyxoviral P proteins.
Sgrignani, J. et al. Molecular determinants for unphosphorylated STAT3 dimerization by integrative modeling

Signal Transducer and Activator of Transcription factors (STATs) are proteins able to translocate into the nucleus, bind DNA and activate gene transcription. STATs proteins play a crucial role in cell proliferation, apoptosis and differentiation. The prevalent view is that STATs proteins are able to form dimers and bind DNA only upon phosphorylation of specific tyrosine residues in the Trans-Activation-domain. However, this paradigm has been questioned recently by the observation of dimers of unphosphorylated STATs (USTATs) by X-ray, FRET and site-directed mutagenesis and a more complex view of the dimerization process and of the dimer role is emerging.
Due to its importance in cancer development and therapy, we focused our study on STAT3. Here we present an integrated modeling study in which we combine the available experimental data with different computational methodologies (homology modeling, protein-protein docking and molecular dynamics), to built reliable atomistic models of USTAT3 dimers. The models were validated performing computational alanine scanning for all the residues at the protein-protein interface. These results confirmed the experimental observations of the importance of some of these residues (in particular Leu78) USTAT3 dimerization process. Moreover, based on these models we were able to predict possible hot-spots (Gln32, Tyr79, Arg84, Arg93) for protein dimerization. In a future perspective our models could be valuable for understating the effects of important pathological mutations at molecular/atomistic level, and for in the design of new inhibitors of dimerization.
Olsson, S., et al. Molecular dynamics of biomolecules through direct analysis of dipolar couplings.


Residual dipolar couplings (RDCs) are important probes in structural biology, but their analysis is often complicated by the determination of an alignment tensor or its associated assumptions. We here apply the Maximum Entropy principle to derive a tensor-free formalism that allows for direct, dynamic analysis of RDCs and holds the classic tensor formalism as a special case. Specifically, the framework enables us to robustly analyze data regardless of whether a clear separation of internal and overall dynamics is possible. Such a separation is often difficult in the core subjects of current structural biology, which include multi-domain and intrinsically disordered proteins as well as nucleic acids. We demonstrate the method is tractable, self-consistent and trivially generalizes to datasets comprised of observations from multiple different alignment conditions.
Associate members: Andrea Cavalli
Status: In progress
Light chain amyloidosis is a disorder associated with aggregation of immunoglobulin (Ig) light chains. Ig light chains with different sequences reveal varied amyloidogenic propensities and it is currently not clear which factors drive fibrillation process and, thus, cause pathological conditions.
In this project, we investigate a repertoire of toxic and non-toxic sequences and perform molecular dynamics simulation for selected light chain models with varied amyloidogenic propensities aiming at the elucidation of molecular determinants of light chain amyloidosis.
Associate members: Andrea Cavalli
Status: In progress
The process of how proteins reach their native basin of structures is poorly understood and constitutes an important problem in molecular biology. This process is called the protein folding problem, and is generally thought to proceed through large, concerted changes in structure.
In this project, we are studying the folding of two small proteins: the WW domain of Pin1 and Porcine peptide YY. These studies are carried out using molecular simulation combined with exact nuclear Overhauser enhancement data and/or chemical shift data obtained at multiple temperatures measured in the groups of collaborators Prof. Riek at the ETH in Zürich or Prof. Zerbe University of Zürich. Specifically, we are integrating all the experimental data with one simulation to obtain a full, thermodynamic and structural description of the protein folding process.
Associate members: Andrea Cavalli
Researchers: Jacopo Sgrignani, Scientist
Status: In progress
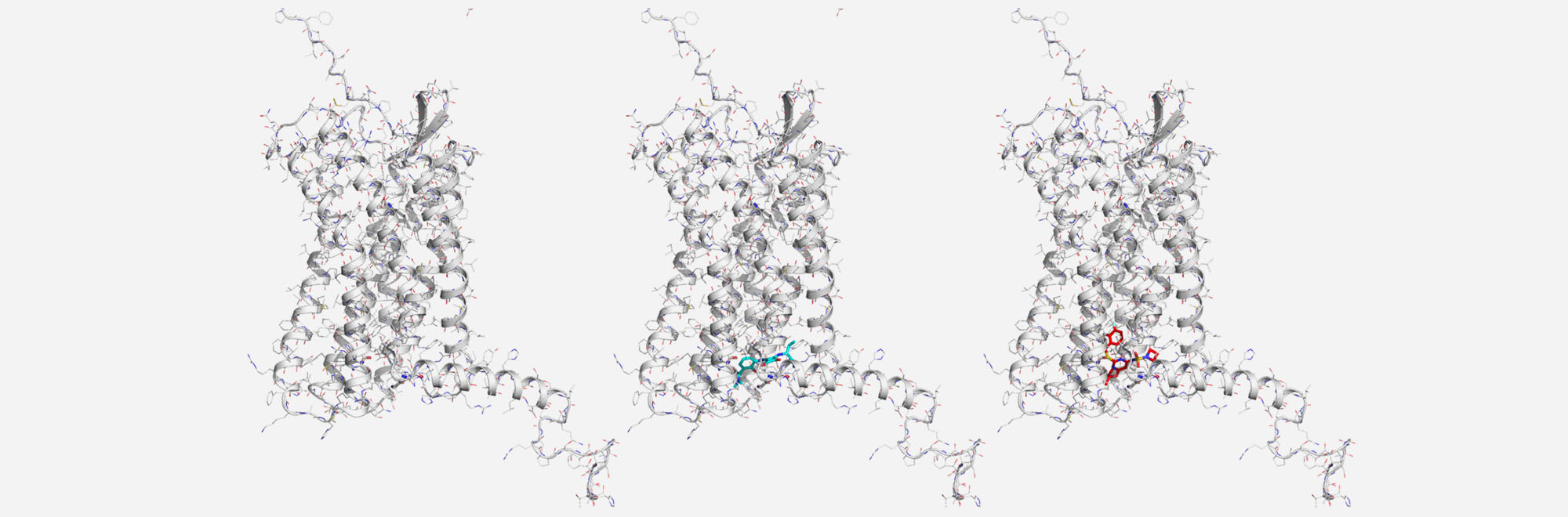
Transcription factors (TFs) are central nodes in multiple oncogenic signalling pathways and represent attractive targets for development of novel cancer treatment strategies. However, very few direct pharmacological inhibitors of transcription factors are currently in the clinical trials. Signal Transducer and Activator of Transcription 3 (STAT3) belongs to the STAT family of transcription factors. As other STAT members, STAT3 is a cytoplasmic protein and is regulated by multiple post-transcriptional modifications (PTM), like phosphorylation, methylation and acetylation.
Increased expression and activity of STAT3 is very common in human cancers. STAT3 has a central role in critical signalling pathways for tumour initiation and progression. STAT3 drives tumour progression by promoting proliferation, survival, metabolic adaptation, tumour angiogenesis and immune tolerance and its downregulation by genetic or pharmacological means prevents or reverts tumorigenesis.
Many anticancer drugs inhibit upstream signaling pathways (e.g., JAK, EGFR) and affect STAT3 activation. In addition to these “indirect” inhibitors of the STAT3 pathway (e.g., JAK inhibitors), there is increasing interest in developing “direct” inhibitors of STAT3 that might interfere with the multiple diverse functions of this TF.
A number of small molecule compounds as well as natural products have been identified as direct STAT3 inhibitors (STAT3i). The aim of this study is to investigate the mechanism of action of a novel class of compounds with STAT3 inhibitory activity. In particular, we will study two compounds that interfere effectively with STAT3 and have potent anticancer activity in various tumor models. Experimental results suggest, that this new class of compounds acts by promoting the formation of large aggregates of STAT3 and that the formation of this aggregates is a direct consequence of conformational changes, disruption of specific inter-domain interactions and partial unfolding of STAT3 induced by STAT3i.
The objective of this study is the characterization of the mechanism of action of STAT3i at a molecular level. In particular we aim to:
Associate members: Andrea Cavalli
Status: In progress
Cyclophilins are a part of the ubiquitous family of enzymes which catalyses the isomerisation transition between peptidyl and prolyl conformations, which plays a crucial role in the folding of many proteins. However, these enzymes have also been identified as a putative drug-target to treat a number of diseases, including viral infections such as Hepatitis C.
In this project we wish to understand the molecular details of the catalysis of Cyclophilin A, and in particular also the inhibition of this function. To this end, we are collaborating with the group Prof. Riek at the ETH in Zürich to analyse high-resolution exact nuclear Overhauser enhancement data and residual dipolar coupling data on Cyclophilin A in complex with cyclosporin, which is known inhibitor of its function, and in absence of this inhibitor.