
Group leader: Luca Varani Researchers: Mattia Pedotti / Technician Research Assistant Status: In progress
Diphtheria is an acute infectious disease caused by the bacterial Diphtheria Toxin (DT). Although mass immunization has virtually eradicated diphtheria from the western world, the disease continues to be a serious health threat in regions like the former USSR, Asia and South America. In the 1990s, for instance, an epidemic caused approximately four thousand deaths in Russia even amongst formerly vaccinated individual, apparently due to a decline in adult immunity level. Beside the medical implications, diphtheria toxin has been extensively characterized at the biochemical level and represents a good model for the study of antibody-toxin interactions. Curiously, however, no structural information on DT-antibody complexes is available so far.
The Lanzavecchia group has isolated a number of human monoclonal antibodies with remarkably strong binding affinity for DT. Some of these antibodies are very potent neutralizers but, intriguingly, they are not those with the stronger binding affinity according to surface plasmon resonance (SPR) measurements. If not binding affinity, what are the determinants of efficient toxin neutralization?
DT is formed by three separated protein domains. ELISA, SPR and cross-competition experiments showed that the best neutralizing antibodies target different regions of the so-called receptor binding domain (figure 4). Perhaps unintuitively, however, we were able to show that the best antibodies do not inhibit interaction of DT with its cellular receptor. We are now trying to determine the mechanism of action of these antibodies through cellular and in vitro experiments. One possibility is that they might block the conformational change required for the activation of DT and subsequent toxicity.
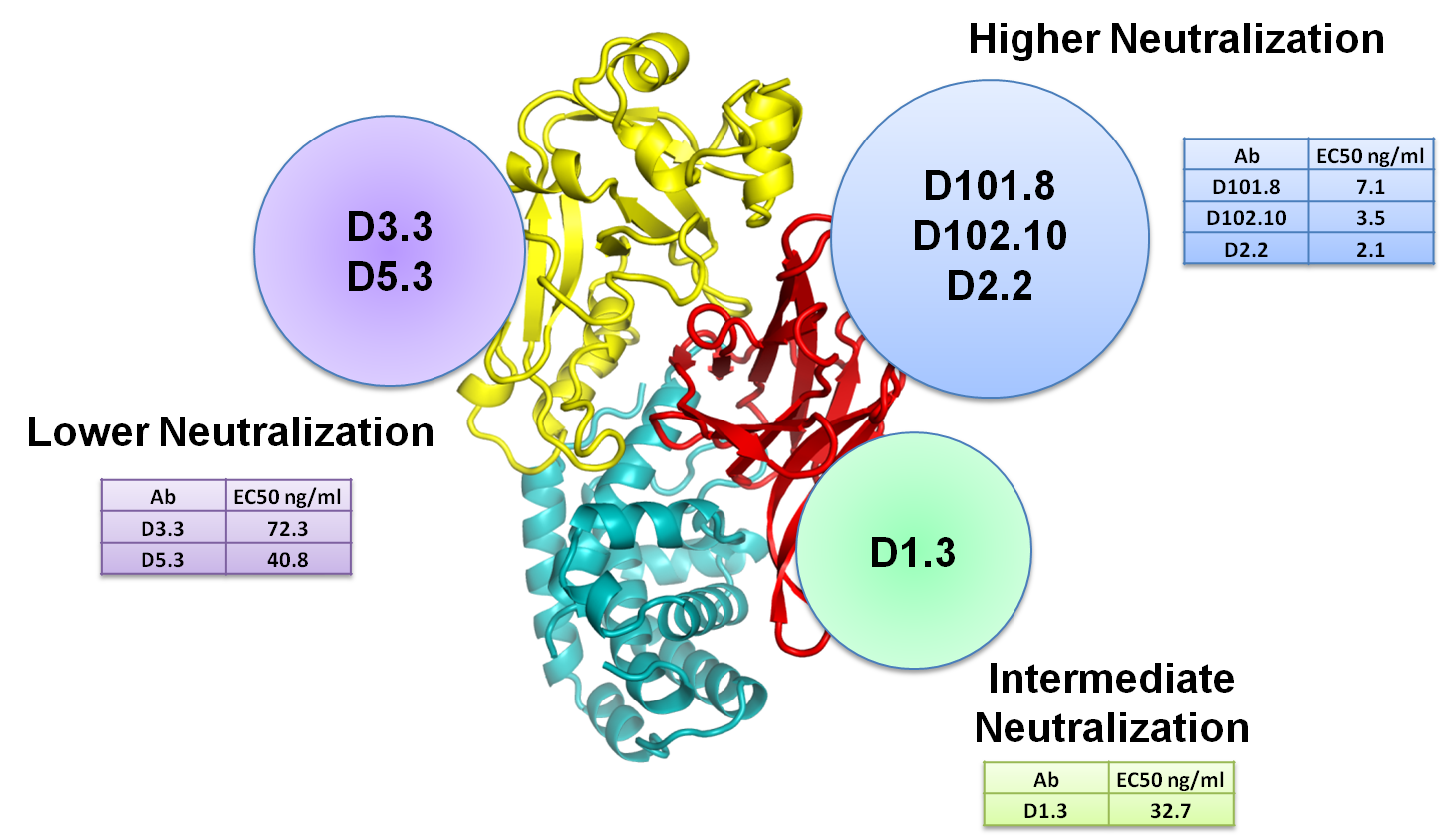
We were able to characterize three groups of human antibodies that bind Diphtheria Toxin (DT) with similar affinity (nanomolar) but have different neutralizing efficacy. Antibodies against the catalytic domain (yellow in the figure) are the worst neutralizer in our panel. Antibodies against the receptor binding domain (red in the figure) show intermediate to high neutralization according to their binding site on this domain.
Group leader: Luca Varani
Researchers: Luca Simonelli / Technician Research Assistant
Status: In progress
Many cancer cells show overexpression of particular proteins that can therefore be targeted by drugs or other therapeutic strategies, at least in theory. In acute myeloid leukemia (AML), a cancer with an high recurrence of relapses due to the presence of Leukemic Stem Cells that are not affected by normal chemotherapic drugs, these leukemic cells differ from normal ones for the strong overexpression of CD123 (IL3 Receptor Alpha) and other proteins such as TIM-3 and CD34 on the cell surface.
We are testing two approaches for selectively targeting AML stem cells with a bio-recognition element capable of discriminating them from normal, healthy cells. Such bio-recognition element could then be linked to either an engineered T-Cell chimeric antigen capable of killing leukemic cells (collaboration with Monza Hospital (IT)) or to nano-vectors capable of delivering drugs or irradiation/heating therapy directly to AML cells (collaboration with EU-Joint Research Center, Ispra (IT)). Previous literature has shown that the approach can succeed in leukemic forms different from AML.
The biggest problem in using overexpressed surface proteins as AML targets is that the bio-recognition element will also target some normal cells that express these proteins at low level. A therapy capable of killing AML cells would, therefore, also kill some normal healthy cells, with deleterious effects. We are attempting to overcome the problem with two complementary strategies. 1) We plan to generate a bi-specific antibody with one antigen binding site capable of recognizing CD123 and a second antigen binding site capable of recognizing TIM3. Both TIM3 and CD123 are expressed on the surface of normal cells but they are not both overexpressed on the same cell as it happens in AML. An antibody that would bind effectively only when it engages both TIM3 and CD123 may be, as a consequence, able to discriminate leukemic from healthy cells. 2) We plan to use either IL3 or an anti-CD123 antibody as bio-recognition element for CD123 (natural receptor alpha for IL3), which is overexpressed in AML cells. In order to avoid unwanted cross-reactivity with healthy cells, we want to investigate the effect of binding affinity on selectivity. Bio-recognition elements with lower binding affinity may engage cells in sufficient number only if their target (e.g. CD123) is abundantly overexpressed, as it is the case in AML cells. In order to tune the binding affinity we are using a combination of experimental methods (solution NMR mapping, mutagenesis, Surface Plasmon Resonance) and computational simulations (docking) to characterize the binding of IL3 and an anti-CD123 antibody to CD123. Visual analysis of the three dimensional structure of CD123 in complex with the aforementioned partners will allow us to design protein mutations with binding affinity lower than the original molecule but still sufficient for successful engagement of leukemic cells.
Group leader: Luca Varani Researchers: Luca Simonelli / Technician Research Assistant Status: In progress
Individuals that survive a viral infection have antibodies (Abs) capable of detecting and neutralizing subsequent attacks by the same virus. These Abs bind antigens (Ags), often viral proteins, through specific atomic interactions between the Ab and the region of the Ag that it recognizes (called epitope). If we understand the structural rules governing Ab-Ag interactions to a given virus, then we have the molecular basis to attempt to design and synthesize new epitopes to be used as vaccines (since most vaccines generate an antibody response) or optimize the antibodies themselves for passive immunization strategies. Comparing the binding of several different antibodies to related Ags should also further our understanding of general principles of recognition.
We recently proposed an experimentally validated computational approach for the rapid and systematic characterization of Ab-Ag complexes. Schematically, we isolate Abs from the blood of human donors infected with a given virus; produce and purify human monoclonal antibodies (in collaboration with A. Lanzavecchia, IRB); characterize their immunological and biophysical properties; determine their epitope through NMR epitope mapping (Figure 1) and use the NMR results to drive and validate computational docking simulations of their complex with the desired antigen. Finally, the structural analysis of the complexes is the starting point for the design of antibody mutations aimed at modifying their properties in a predictable manner, with the goal of validating our results and engineer new antibodies with improved properties.
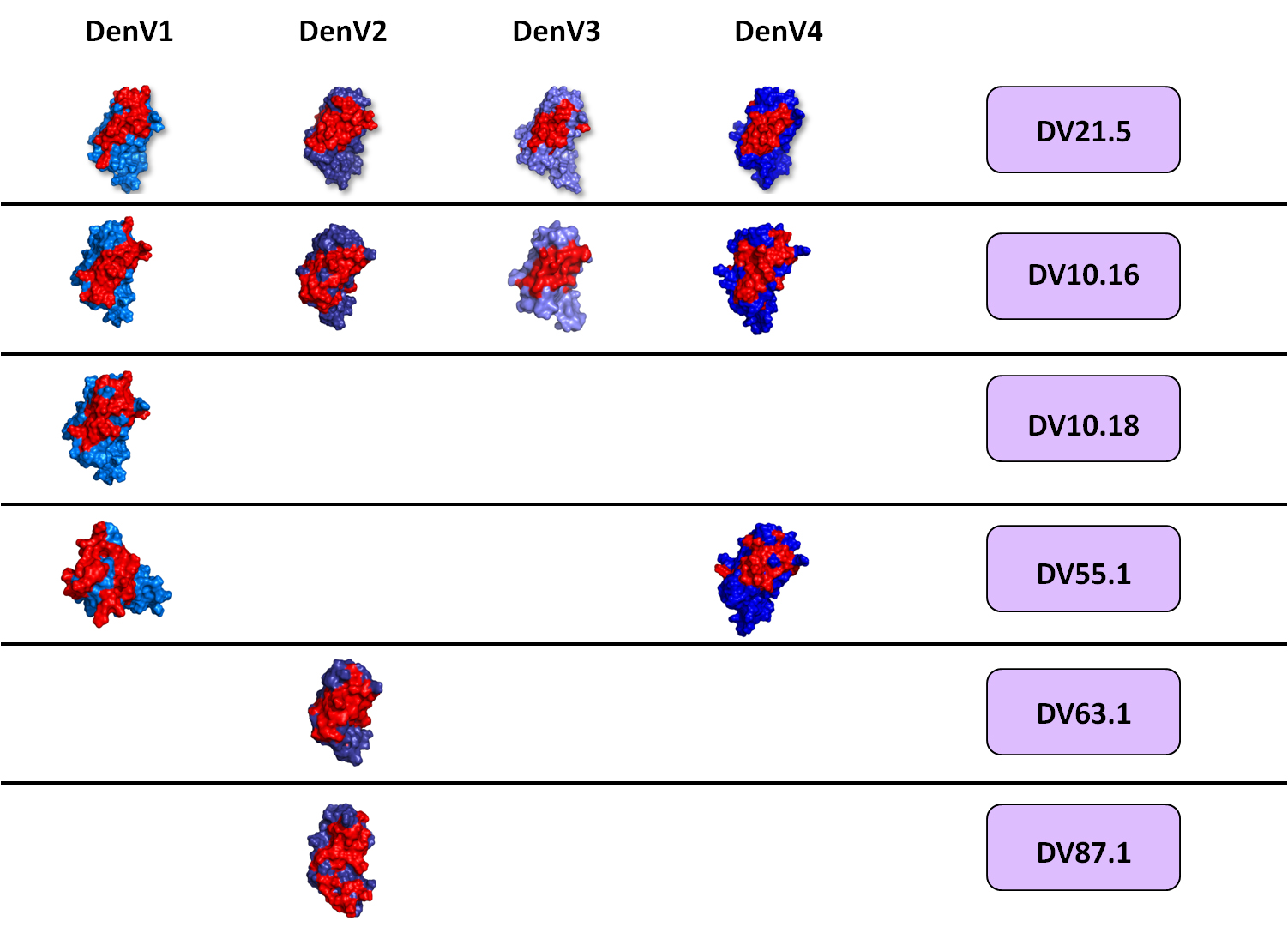 Figure 1: The epitope of several antibodies against Dengue was mapped with solution NMR spectroscopy. Epitopes are shown in red over the surface representation of the so-called DIII, a part of the protein forming the surface of Dengue virus and the site recognized by most neutralizing antibodies against Dengue. Each line indicates a different antibody; the vertical columns correspond to the four main Dengue serotypes.
Figure 1: The epitope of several antibodies against Dengue was mapped with solution NMR spectroscopy. Epitopes are shown in red over the surface representation of the so-called DIII, a part of the protein forming the surface of Dengue virus and the site recognized by most neutralizing antibodies against Dengue. Each line indicates a different antibody; the vertical columns correspond to the four main Dengue serotypes.
Dengue Virus: a case study
Dengue Virus (DENV) is a flavivirus responsible for 100 million annual human cases, including 500,000 hospitalizations and 20,000 deaths with an economic burden rivaling that of malaria. Although DENV has been mainly restricted to the tropical region, both its epidemic activity and its geographic expansion are increasing as travel, urbanization and climate changes create favorable conditions for vector and virus dissemination. An estimated 2.5 billion people are at risk of infection.
No cure or vaccine for DENV is currently available. The effort to find one has been hampered by the presence of four different dengue serotypes (DENV1–4) and by a poorly understood process almost unique in human medicine: antibody-dependent enhancement (ADE). Abs raised against a previous Dengue infection facilitate subsequent infection by a different serotype and lead to dengue hemorrhagic fever, an often lethal form of the disease. This feature complicates the task of finding a vaccine, since a vaccine that would not protect equally against all four serotypes would actually contribute to the emergence of dengue hemorrhagic fever.
At the structural level, the most interesting region to study is the so-called Domain III of Dengue E protein (DIII), which forms the surface of the virus. DIII is the main target of neutralizing antibodies against DENV and it is relatively small, making it ideal for NMR and computational studies.
Our aim is to compare several antibodies bound to DIII of the four Dengue serotypes, searching for correlations between immunological and structural trends and exploiting them to further our understanding of antibody-antigen interaction and ADE, as well as a basis for drug design and improved vaccine strategies. In a simplistic example, should we find that all Abs effective against DENV4 have a positive charge in a particular three-dimensional position, we would try to introduce such a charge in Abs lacking it, thus improving their characteristics. Conversely, should all effective Abs against a certain serotype recognize a particular epitope, then it is conceivable to prepare an antigen sharing the best epitopes of each serotype as a possible vaccination agent.
This work was done in collaboration with Antonio Lanzavecchia and Federica Sallusto, IRB.
Rational antibody engineering to better neutralize DengueWe determined the binding site of monoclonal antibody DV32.6 on DIII of all Dengue serotypes with NMR epitope mapping and used this information to guide and validate computational docking simulations yielding the three-dimensional structure of the antibody/antigen complexes. Visual analysis of such structures allowed us to design antibodies with altered binding sites that could: A) avoid binding to all serotypes. B) Bind only to one or two serotypes; eliminating unwanted cross-reactivity is a useful endeavor for therapeutic antibodies or when designing bio-recognition elements. C) Neutralize Dengue virus serotype 1 40 times more effectively than the original antibody or neutralize all serotypes between 10 and 17 fold better than the original molecule.
Overall, 18 out of 22 point mutations that we designed had the effect predicted by the computational models. The work proves that experimentally validated computational docking is an accurate, rapid and powerful tool for the characterization and rational engineering of antibodies (Figure 2).
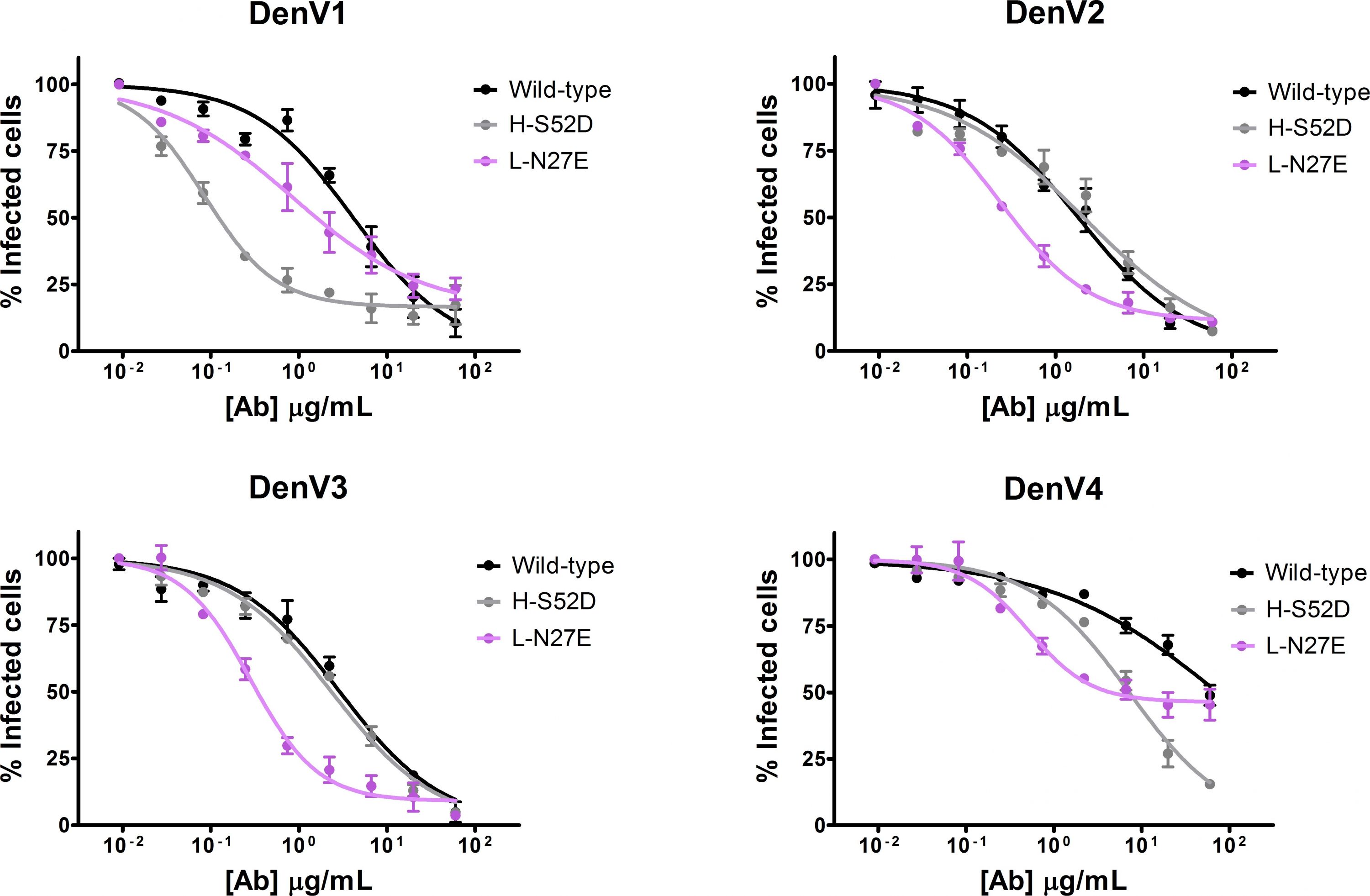 Figure 2: We designed two antibody mutants with the intent of improving its neutralization properties. H-S52D (gray) neutralizes DenV1 40 times more efficiently than the wild-type (black) and L-N27E (violet) is better than the wild-type in all serotypes, albeit to a lesser extent. Viral neutralization assays are shown; the amount of infected cells (y axis) decreases at increasing antibody concentration (x axis). In comparison to the wild-type, a lower concentration of mutants is required to neutralize the same amount of virus.
Figure 2: We designed two antibody mutants with the intent of improving its neutralization properties. H-S52D (gray) neutralizes DenV1 40 times more efficiently than the wild-type (black) and L-N27E (violet) is better than the wild-type in all serotypes, albeit to a lesser extent. Viral neutralization assays are shown; the amount of infected cells (y axis) decreases at increasing antibody concentration (x axis). In comparison to the wild-type, a lower concentration of mutants is required to neutralize the same amount of virus.
Structural Characterization of a potent neutralizer of Dengue Virus
DV87.1 is probably the most potent Dengue antibody described so far in the literature. It binds the surface protein of Dengue virus with nanomolar affinity and has an EC50 of 0.004µg/ml.
We used solution NMR spectroscopy to define its epitope on Dengue serotype 2 (figure 3) and then used this information to validate computational simulations aimed at obtaining the three-dimensional structure of the antibody in complex with its antigen. The antibody binds to an epitope not before described for other Dengue antibodies. Its position in the structure suggests that DV87.1 may neutralize Dengue Virus by blocking the conformational changes required for membrane fusion and infection of the host cells. Experiments are undergoing to obtain direct evidence on this hypothesis.
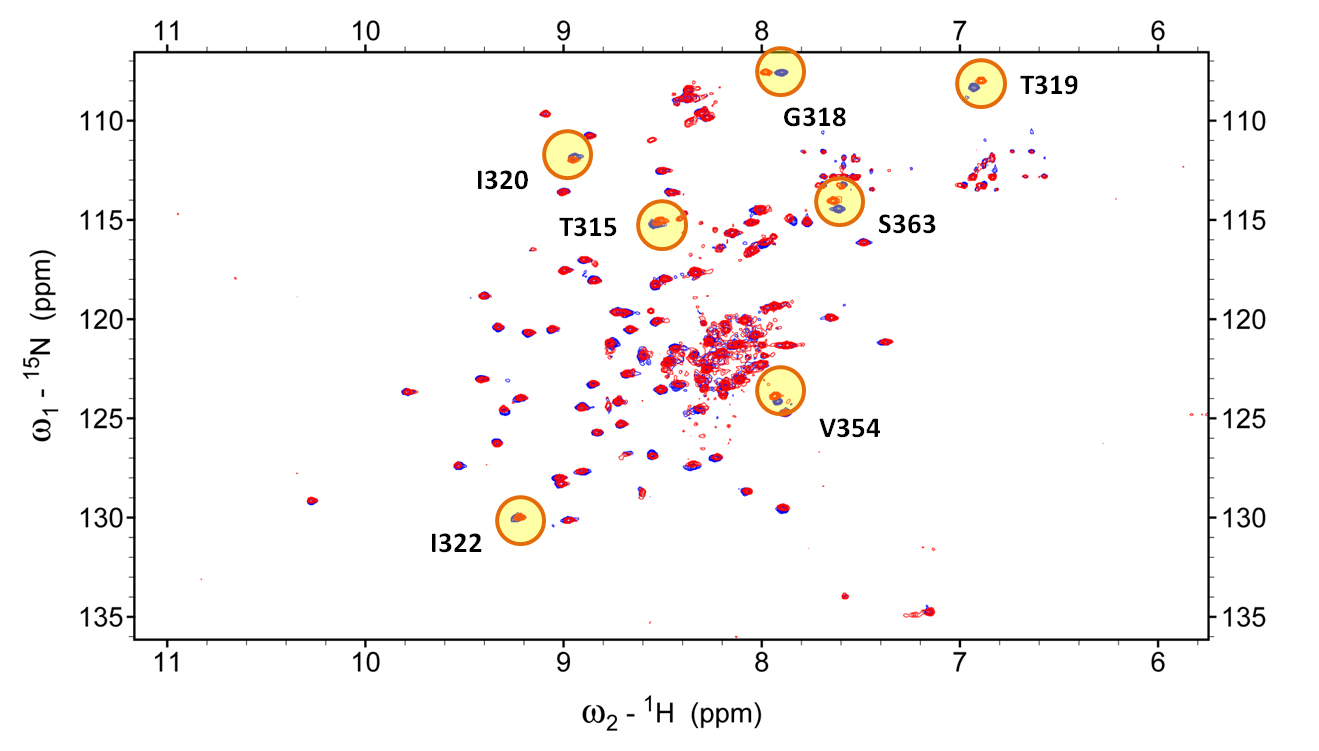 Figure 3: NMR spectrum of the Dengue antigen (DIII of DenV2) by itself (blue) and in complex with antibody DV87.1 (red). In this type of experiment, each peak corresponds to an individual antigen residue. Differences in the position of the peaks between free and bound spectrum allow us to determine which residues are affected by antibody binding and are, thus, at the interface (epitope). Some of the residues whose signal differs between free and bound form are indicated in yellow.
Figure 3: NMR spectrum of the Dengue antigen (DIII of DenV2) by itself (blue) and in complex with antibody DV87.1 (red). In this type of experiment, each peak corresponds to an individual antigen residue. Differences in the position of the peaks between free and bound spectrum allow us to determine which residues are affected by antibody binding and are, thus, at the interface (epitope). Some of the residues whose signal differs between free and bound form are indicated in yellow.Group leader: Luca Varani
Researchers: Mattia Pedotti / Technician Research Assistant
Status: In progress
The FP7 research consortium RADAR aims at developing label-free biosensor platforms for the monitoring of organic pollutants in the environment and for the surveillance of industrial production processes. The so-called Estrogen Disrupting Compounds (EDCs) are a class of pollutants capable of binding to human and animal estrogen receptors and disrupting their normal function. EDCs are known to be responsible for diseases ranging from cancer to sex changes in aquatic animals. Thousands of different EDCs exist, many of which still unknown. The concept behind the RADAR project is simple: EDCs exert their adverse function by binding to the Estrogen Receptor protein (ER). By using the ER as a bio-recognition element we can detect all compounds capable of binding to it and, therefore, potentially harmful.
Our role in the RADAR consortium is to design and produce a rationally modified ER that can 1) be easily attached to a sensor surface (via chemical tags); 2) be produced at low cost (E.Coli); 3) have an altered binding selectivity and increased affinity for selective organic compounds, so that a binding event would signal the presence of a particular class of compounds.
Structural analysis and computational docking allowed us to design a mutated ER capable of binding bis-phenolic compounds with increased activity in comparison to the wild type protein. This mutated ER has been successfully produced, stabilized for a period of several months and attached to the surface of a label-free biosensor platform developed by the RADAR consortium.